Repurposing bumetanide for Alzheimer’s


|
# # # # Bumetanide (Bumex) is a diuretic drug (a medication that removes water, by increasing the production of urine). It is used to treat swelling caused by heart failure or liver or kidney disease. It is a widely used drug that has been well characterised in clinical use. Recently researchers conducted a screening study to identify clinically available agents that might be useful in the treatment of the cognitive decline associated with a genetic risk factor for Alzheimer’s: APOE4 The top drug identified in their study was bumetanide. In today’s post we will discuss what APOE4 is, we will review the results of the new study, and we will look at why these findings could be interesting for Parkinson’s. # # # # |
 Source: Pharmacysafety
Source: Pharmacysafety
Many years ago, I was at a patient-research interaction event and a world-leading genetics researcher was asked by someone in the audience if they had had their DNA sequenced.
They said ‘no‘.
The person asking the question frowned and asked ‘why not? You have all the technology and knowledge – don’t you want to know more about yourself?‘
The researcher replied “No. Having your DNA sequenced should not be taken lightly. You might learn stuff about yourself that you don’t want to know”
They used the example of possibly being an APOE4 carrier (who have a higher risk of cognitive decline during aging). The geneticist declared that they would rather not know that kind of information for fear of the impact that it could have on their life.

The questioner respected the honest answer and the conversation that followed was really interesting. More recently, however, as we have learned more about APOE4 and new drugs are being targeted at this risk factor, I have often wondered if their decision would still stand. Are we approaching an age when we might want to know if we are APOE4 carriers?
Hang on a moment. What is this APOE4 thing?
Apolipoprotein E4 (also known as apoE4) is the most prevalent genetic risk factor of Alzheimer’s. It is present in more than half of Alzheimer’s cases.
What is ApoE4?
Apolipoprotein E is a protein that is involved in the transportation of lipids and injury repair in the brain.
Outside of the brain, ApoE is primarily produced by the liver and macrophage cells in the blood. It functions by mediating cholesterol metabolism.
The apolipoprotein E gene (the region of DNA that provides the instructions for making APOE protein) sits on chromosome 17 of our DNA.
Hang on a second. What happened to the “4” in ApoE4? Why did you shift from ApoE4 to ApoE?
Because ApoE is the gene, but it is “polymorphic“.
And what does that mean?!?
Polymorphic means that there are two or more clearly different morphs (or versions/forms) of the gene due to variations.
So everyone doesn’t have the same ApoE gene?
No, there are three common versions/forms (or morphs) of the gene:
- ApoE2,
- ApoE3
- ApoE4
These different forms of the ApoE gene result in tiny differences in the ApoE protein (only one or two amino acids at positions 112 and 158). But these differences alter the activity of the protein and how they interact with the world.
Now you may remember that everyone inherits two copies (or alleles) of a gene, one from each parent. Having two copies of each gene is mother nature’s insurance policy that an organism will have an increased chance of surviving. But sometimes those two copies of the same gene can vary slightly.
Therefore it is possible for you to have two different versions of the ApoE gene. You could have any one of the following six combinations (or “genotypes”):
- E2/E2 (which occurs in 1% of the UK population)
- E2/E3 (11%)
- E2/E4 (2%)
- E3/E3 (61%)
- E3/E4 (23%)
- E4/E4 (2%)
Individuals carrying any combination of the ApoE4 gene are at increased risk of developing Alzheimer’s, compared with people carrying the more common ApoE3 gene or the rarer ApoE2 gene (the ApoE2 gene actually decreases risk).
 Source: PMC
Source: PMC
And before we go on it is important to understand that these genetic variations are simply an “association” with increased risk. They do not necessarily mean that someone with two copies of ApoE4 will definitely develop Alzheimer’s. That individual may not.
In addition to these ApoE4 variations there are probably dozens of counter balancing genetic variants that may reduce the risk of Alzheimer’s. Our knowledge of the genetics of these conditions still requires a lot of work.
Got it. Are researchers trying to find treatments that reduce the impact of ApoE4?
Yes they are.
And this week an interesting report was published:
 Title: Experimental and real-world evidence supporting the computational repurposing of bumetanide for APOE4-related Alzheimer’s disease
Title: Experimental and real-world evidence supporting the computational repurposing of bumetanide for APOE4-related Alzheimer’s disease
Authors: Taubes A, Nova P, Zalocusky KA, Kosti I, Bicak M, Zilberter MY, Hao Y, Yoon SY, Oskotsky T, Pineda S, Chen B, Aery Jones EA, Choudhary K, Grone B, Balestra ME, Chaudhry F, Paranjpe I, De Freitas J, Koutsodendris N, Chen N, Wang C, Chang W, An A, Glicksberg BS, Sirota M, Huang Y.
Journal: Nature Aging volume 1, pages 932–947 (2021)
PMID: N/A
In this report, the investigators screened 1,300 drugs to identify those that had the best potential to reverse APOE genotype-specific features (based on computational analysis of the transcriptional signatures of the protein and also the drugs being screened).
This screen identified bumetanide as the top candidate.
What is Bumetanide?
Bumetanide is a medication that is used to treat heart failure.
Heart failure (sometimes referred to as congestive heart failure) occurs when the heart is unable to pump sufficiently enough to maintain the required blood flow to meet the body’s needs. The most common causes of heart failure include coronary artery disease, high blood pressure, atrial fibrillation, valvular heart disease, and lifestyle issues (such as excess alcohol use). Overall around 2% of adults have heart failure; in those over the age of 65, this percentage increases to 6–10%. In 2015, it was estimated to affected approximately 40 million people worldwide (Source).
Common symptoms include:
- shortness of breath
- excessive tiredness
- leg swelling
A common treatment option for heart failure are diuretics, and bumetanide is a diuretic.
What are diuretics?
Diuretics (sometimes called water pills) are medications that have been designed to increase the amount of water and salt expelled from the body as urine.
There are three types of diuretic medications. They are:
- Thiazide
- Loop
- Potassium-sparing
Thiazide diuretics are the most commonly prescribed, generally for the treatment of high blood pressure. This class of drugs not only decreases the level of fluids in your body, they also cause your blood vessels to relax. Potassium-sparing diuretics reduce fluid levels in your body without – as the label suggests – causing you to lose potassium. The other types of diuretics can cause you to lose potassium, which can result in other health complications such as arrhythmia.
And then there are loop diuretics, which also decrease the level of fluid in the body. They achieve this by interfering with the transportation of salt and water across particular cells in the kidneys.
Those cells are located in a structure called the loop of Henle. Hence the name: Loop diuretic.
Bumetanide is a loop diuretic.
Ok, so what did the researchers do next?
Next, the researchers evaluated the ability of bumetanide to rescue neuronal deficits in mice that were genetically engineered to carrying human APOE4. These mice develop cognitive issues as they age, but the investigators found that bumetanide treatment was able to prevent this, restoring normal spatial learning in the mice.
 Source: Pinterest
Source: Pinterest
The researchers also reported that bumetanide treatment improved the health of human neurons (that carried APOE4) grown in cell culture. And as a final investigation, they looked at two indpendent medical record databases and found (in both) that bumetanide exposure was associated with a significantly lower prevalence of Alzheimer’s in individuals over 65 years of age.
Encouraged by their results, the investigators concluded by saying the obviously bumetanide warrants further assessment in terms of APOE4 biology. It has been proposed that the data “make a good case to conduct a proof-of-concept trial of bumetanide in people with genetic risk” (Source).
|
# RECAP #1: APOE4 is a genetic risk factor for Alzheimer’s and cognitive issues in aging. Researchers have been trying to identify agents that can influence the outcomes for carriers of this genotype. A recent screening study has identified the heart failure treatment bumetanide as a candidate for potential pharmacological intervention against the effects of APOE4. # |
Could this research on APOE4 be relevant to Parkinson’s?
Yes.
APOE4 is a problem not only in Alzheimer’s, but also Parkinson’s. Carriers of an APOE4 allele who develop Parkinson’s typically have an earlier onset of the condition and there is a higher risk developing cognitive issues (Click here to read more about this). In fact, irrespective of neurodegenerative condition, APOE4 carriers have lower performance in cognitive measure compared to non-APOE4 carriers (Click here to read more about this).
And there is evidence in Parkinson’s that APOE4 could be directly influencing the progression of the condition. Early last year two research groups published back-to-back reports (meaning that they followed each other in the same journal) supporting this idea.
Here is the first report:
 Title: APOE genotype regulates pathology and disease progression in synucleinopathy.
Title: APOE genotype regulates pathology and disease progression in synucleinopathy.
Authors: Davis AA, Inman CE, Wargel ZM, Dube U, Freeberg BM, Galluppi A, Haines JN, Dhavale DD, Miller R, Choudhury FA, Sullivan PM, Cruchaga C, Perlmutter JS, Ulrich JD, Benitez BA, Kotzbauer PT, Holtzman DM.
Journal: Sci Transl Med. 2020 Feb 5;12(529). pii: eaay3069.
PMID: 32024799 (This report is OPEN ACCESS if you would like to read it)
In this study, the investigators the researchers genetically engineered mice with the A53T mutation in the alpha synuclein gene.
What does that mean?
On this website we are forever talking about a protein called alpha synuclein.
It sounds like a distant galaxy, but it is one of the most common proteins in our brains. It makes up about 1% of all the protein in a neuron. When alpha synuclein protein is produced by a cell, it normally referred as a ‘natively unfolded protein’, in that is does not really have a defined structure. When it is first produced, alpha synuclein will look something like this:

Alpha synuclein. Source: Wikipedia
In this form, alpha synuclein is considered a monomer – which is a single molecule that can bind to other molecules. When it does bind to other alpha synuclein proteins, they form an oligomer (a collection of a certain number of monomers in a specific structure). It is believed that alpha synuclein has certain functions as a monomer, but may also have specific tasks as a oligomer.
In Parkinson’s, alpha synuclein also binds (or aggregates) to form what are called ‘fibrils’.

Microscopic images of alpha synuclein (AS) monomers, oligomers and fibrils. Source: Brain
And it is believed that the oligomer and fibril forms of alpha synuclein protein that aggregate together, and then go on to form what we call Lewy bodies.
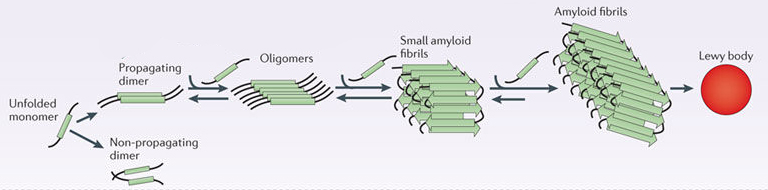 Parkinson’s associated alpha synuclein. Source: Nature
Parkinson’s associated alpha synuclein. Source: Nature
What are Lewy bodies?
Lewy bodies are dense circular clusters of aggregated alpha synuclein protein (and other proteins) that are found in specific regions of the brain in people with Parkinson’s (Click here for more on Lewy bodies).
Lewy bodies are one of the cardinal features of the Parkinsonian brain – they are used to help make postmortem diagnoses.
 A cartoon of a neuron, with the Lewy body indicated within the cell body. Source: Alzheimer’s news
A cartoon of a neuron, with the Lewy body indicated within the cell body. Source: Alzheimer’s news
Ok, but what is the “A53T mutation” part all about?
The section of DNA that gives rise to alpha syncuclein protein is called SNCA.
And there are several genetic variations inside of SNCA that are associated with an increased risk of developing Parkinson’s. A53T is the name of one of those genetic variations.
As you can see in the image below, A53T lies in the red (Amphipathic) region of SNCA along with several other genetic variants, such as A30P and E46K:

Mice have been genetically engineered to carry the human SNCA gene with the A53T genetic mutation (Click here to read the original report). These mice initially exhibit hyperactivity and then start to display signs of alpha synuclein protein accumulation and aggregation at about four to six months of age. They also pass away earlier than normal mice (12-14 months of age, compared to 20+ months for normal mice).
I see. So what did the researchers do with the mice?
In addition to the A53T mutation in the alpha synuclein gene, the investigators also engineered the mice so that they carried either no ApoE gene or one of the 3 human APOE variations (ApoE2, ApoE3, or ApoE4).
They then bred these “mutant alpha synuclein+ApoE variant” mice and watched to see what happened. At 12 months of age, the researchers found that the mice carrying the ApoE4 version of the ApoE gene had higher levels of alpha synuclein than the rest of the mice.
In fact, the ApoE2 mice had undetectable levels of alpha synuclein. In addition, the ApoE2 mice survived longer, while the ApoE4 mice died ealier. The ApoE4 mice also had more motor problems than the ApoE2 mice.
On top of the mouse research, the investigators also looked at two independent clinical cohorts of people with Parkinson’s and they found that individuals with two copies of the ApoE4 gene exhibited faster rates of cognitive decline compared with people with Parkinson’s who do not have a copy of ApoE4.
And the second study found very similar results:
 Title: APOE4 exacerbates α-synuclein pathology and related toxicity independent of amyloid.
Title: APOE4 exacerbates α-synuclein pathology and related toxicity independent of amyloid.
Authors: Zhao N, Attrebi ON, Ren Y, Qiao W, Sonustun B, Martens YA, Meneses AD, Li F, Shue F, Zheng J, Van Ingelgom AJ, Davis MD, Kurti A, Knight JA, Linares C, Chen Y, Delenclos M, Liu CC, Fryer JD, Asmann YW, McLean PJ, Dickson DW, Ross OA, Bu G.
Journal: Sci Transl Med. 2020 Feb 5;12(529). pii: eaay1809.
PMID: 32024798 (This report is OPEN ACCESS if you would like to read it)
In this study, the researchers used viruses to deliver alpha synuclein to mice.
How did they do that?
Viruses are simple things.
They are a basically a shell with a bit of DNA inside it. The shell attaches to a cell, injects its DNA and the cell then produces more copies of the virus. Very simple, right?
Now, if you take the viral DNA out of the shell of the virus, what you have left is a very effective biological delivery system – one which can be used to delivery any kind of DNA you want.
Researchers have embraced this idea and used viruses as research tools. And in the current study, the investigators used viruses to deliver alpha synuclein DNA (called SNCA) into the brains of mice which carried either the ApoE2, ApoE3, or ApoE4 versions of ApoE.
They found that mice carrying ApoE4 had worse alpha synuclein pathology, and more behavioral problems than mice with ApoE2 or ApoE3.
And these researchers also looked at humans, but they focused their analysis on postmortem brain samples. They assessed levels of alpha synuclein pathology in section of brain from people who passed away with Lewy Body Dementia (Click here to read a previous SoPD post about this topic). Specifically, the researchers compared samples from people carrying ApoE4 vs non-carriers, and they reported more severe alpha synuclein pathology in the ApoE4 carriers.
Both groups of researchers concluded their studies by suggesting that the version of ApoE each of us carries can regulate alpha synuclein pathology.
What does this mean if I am an ApoE4 variant carrier?
As we discussed above, these results are based on an association, and not all individuals with ApoE4 variants go on to develop Alzheimer’s (and the case is most likely the same with alpha synuclein pathology). So there is no reason to immediately start panicking.
All of this research needs to be further explored and expanded on.
|
# # RECAP #2: APOE4 has been shown to be relevant to Parkinson’s as well as Alzheimer’s. Data published last year indicates that APOE4 carriers have a more rapid progression of the condition. Mice carrying human ApoE4 had worse alpha synuclein pathology, and postmortem human brain samples from APOE4 carriers with Lewy Body Dementia exhibited more severe alpha synuclein pathology. # # |
Given the APOE4 associations, has bumetanide been tested in the context of Parkinson’s?
Yes it has.
In 2018, this study was published:
 Title: GABAergic inhibition in dual-transmission cholinergic and GABAergic striatal interneurons is abolished in Parkinson disease
Title: GABAergic inhibition in dual-transmission cholinergic and GABAergic striatal interneurons is abolished in Parkinson disease
Authors: Lozovaya N, Eftekhari S, Cloarec R, Gouty-Colomer LA, Dufour A, Riffault B, Billon-Grand M, Pons-Bennaceur A, Oumar N, Burnashev N, Ben-Ari Y, Hammond C.
Journal: Nat Commun. 2018 Apr 12;9(1):1422.
PMID: 29651049 (This research article is OPEN ACCESS if you would like to read it)
In this study, the researchers found that half of the cholinergic interneurons in the striatum are both cholinergic and GABAergic. They also found that the dopamine depletion observed in Parkinson’s, results in these cholinergic/GABAergic interneurons becoming dysfunctional, and enhancing the inhibitory effect in the striatum. But more importantly, the investigators found that bumetanide rescued this effect.
Cool huh!
Umm, ok. But what does any of that actually mean?
Let’s start at the beginning.
The striatum is a structure deep in the brain. It is broken into two subregions in humans, called the putamen and the caudate nucleus. The dopamine-producing neurons of the substantia nigra (these are the cells that are badly affected in Parkinson’s) have long projections (or axons) that extend up into the brain to the putamen and caudate nucleus, and this is where the cells release the bulk of their dopamine.

The projections of the substantia nigra dopamine neurons. Source: MyBrainNotes
The striatum is made up of different types of neurons.
The vast majority of the cells in the striatum are called medium spiny neurons (also known as spiny projection neurons). They represent 95% of neurons within the human striatum, and they are a type of inhibitory cell. they produce a chemical called gamma–Aminobutyric acid (or just GABA).

GABA. Source: Wikipedia
GABA is a neurotransmitter. A neurotransmitter is a chemical that is used to pass a signal from one neuron to another. Dopamine – the chemical that is severely reduced in the Parkinsonian brain – is also a neurotransmitter.
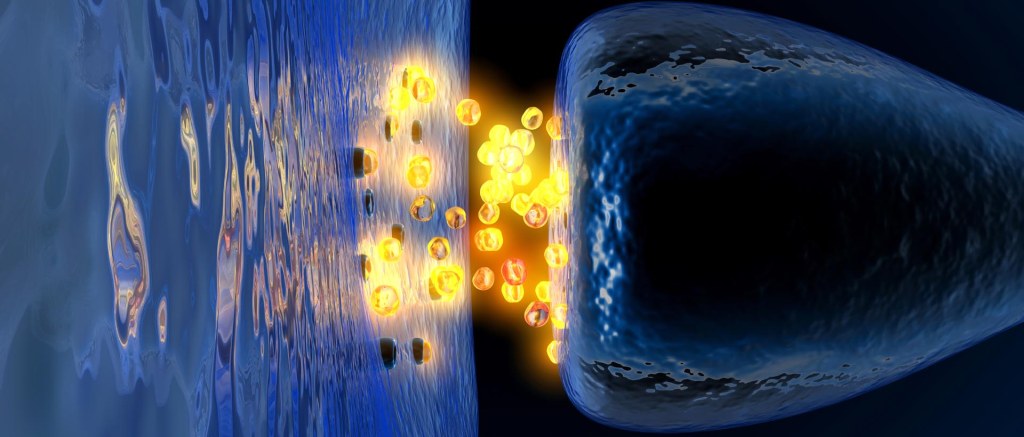
A neurotransmitter being released by one cell (right) and binding to another. Source: Truelibido
Another population of cells in the striatum are the cholinergic interneurons. They make up only about 1–2% of all the cells in the striatum, but they are very large cells which send out dense networks of branches throughout the striatum, allowing them to communicate with lots of other cells. Cholinergic interneurons produce a neurotransmitter called acetylcholine.
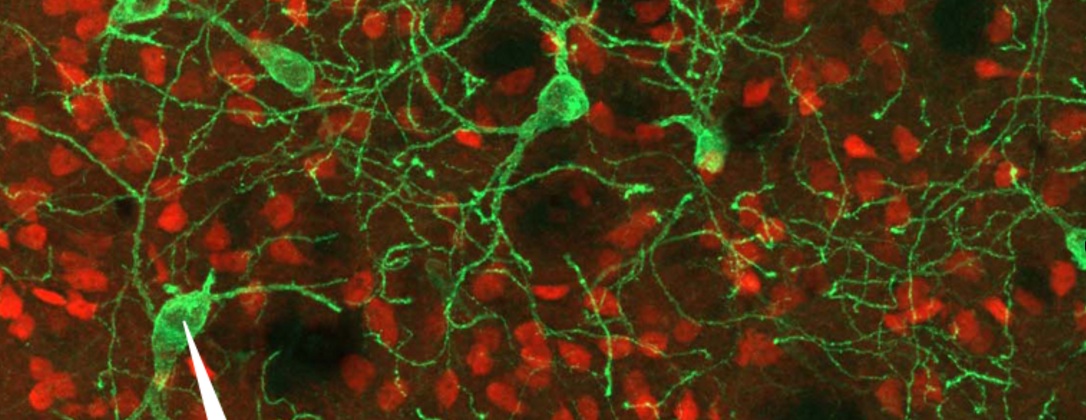
Cholinergic neurons (green) and GABA neurons (red) in the striatum. Source: Neuro-marseille
Now the researchers in France who published the report we are reviewing today found that half of the cholinergic interneurons in the mouse striatum produce both acetylcholine and GABA. In the image below, note the white arrow pointing at the red cell (which is stained with a dye labelling the enzyme ChAT – which is required for the production of acetylcholine). That cell is also producing an enzyme called GAD 65 (white arrow in the middle panel), which is required for the production of GABA.

Example of a cholinergic GABA-producing interneuron. Source: Nature
The idea that neurons can produce and release two different neurotransmitters is not unique in the brain and it has been previously reported (Click here for an example). The researchers referred to these cholinergic GABA-producing interneurons in the striatum as CGINs.
When these CGIN neurons released the neurotransmitter acetylcholine, it was found to cause an excited response in neighbouring cells, while the GABA that was released was found to be inhibitory. The researchers suggested that these CGINs (or more importantly the balance between cholinergic and GABAergic activity from these cells) play a crucial role in the synchronisation of striatal networks, and they next wanted to look at what happens in the absence of dopamine – as in the case of Parkinson’s.
In Parkinson’s, the branches (or axons) of the dopamine neurons that extend up to the putamen and caudate nucleus gradually disappear as the dopamine neurons of the substantia nigra are lost. When one looks at brain sections of the putamen after the axons have been labelled with a dark staining technique, this reduction in axons is very apparent over time, especially when compared to a healthy control brain.

The putamen in Parkinson’s disease (across time). Source: Brain
EDITOR’S NOTE: I WOULD JUST LIKE TO ADD THAT THE IMAGE ABOVE IS NOT REPRESENTATIVE OF EVERYONE WITH PARKINSON’S. THE IMAGE IS BEING USED HERE TO PROVIDE AN EXAMPLE OF THE DOPAMINE FIBRE LOSS OBSERVED IN THE PUTAMEN. THIS PROCESS CAN TAKE LONGER IN SOME INDIVIDUALS THAN THE PERIOD OF TIME INDICATED.
In the absence of dopamine, the striatum gradually puts out a more and more inhibitory signal (blocking a person’s ability to move normally). This results in a very inhibitory signal being sent from the brain, down the spinal cord to the muscles of the body. So rather than having a carefully balanced combination of excitatory and inhibitory signals, the brain is sending out signals that inhibit more than encourage movement. Hence the slowness of movement witnessed in Parkinson’s.

Weakened excitatory signals (green) and enhanced inhibitory signals (red) in the Parkinsonian brain. Source: Animal Physiology 3rd Edition
So what happened to the CGIN neurons in the absence of dopamine?
The French researchers found that these CGIN neurons lost their ability to respond to GABA.
When the investigators treated the CGIN neurons of normal mice with a GABA receptor agonist – that is a molecule that stimulates the GABA receptor and inhibits cell activity (we have previously discussed agonists – click here to read more) – the CGIN cells activity reduced. But in mice that had been treated with a neurotoxin (6-OHDA) that killed the dopamine neurons, the GABA receptor agonist had no effect on the CGIN cells activity.
The CGIN neurons had lost their ability to respond to GABA.
This effect meant that the careful balance between cholinergic and GABAergic activity from these cells was completely thrown out of whack, leaving the CGIN cells continuously sending out an inhibitory signal. And this constant inhibitory signal, left the striatum even more inhibited.
If none of this is making any sense, understand one thing: the researchers identified a drug that could correct this situation (both in cell culture and in mice).
Was that drug bumetanide – the drug you mentioned above?
Yes, it was.
But how does it fix the situation?
Bumetanide functions as an NKCC1 chloride importer antagonist.
And what on earth does that mean?!?
Ok, so chloride is an essential electrolyte.
And before you ask: An electrolyte is a substance that dissociates into ions in solution and acquires the capacity to conduct electricity. In neurons, this property is essential for maintaining cell homeostasis and transmitting action potentials in neurons.
An action potentials is a critical part of how one neuron passes a signal on to another neuron.
The action potential is an electrical impulse that is passed down the branch (or axon) of a neuron to the axon terminals where it stimulates the release of chemical messengers to pass the signal to the next neuron.

Source: KhanAcademy
When a neuron is at rest, there is a high concentration of sodium (Na+) ions and chloride (Cl-) ions outside of the neuron (in the extracellular fluid) compared the situation inside the cell (the intracellular fluid) where there is a high concentration of potassium (K+) ions.

Source: Washington
During an action potential, as the impulse is moving along the axon, this balance is reversed – sodium ions and chloride ions rush inside of the neuron, while potassium moves out. After the impulse has passed, there are active mechanisms that return the balance to normal (high concentration of sodium ions and chloride ions outside of the cell, etc).
This video will explain to you what an action potential is:
Now, an important feature of chloride is that while it is technically involved with action potentials, it is not actually essential for an action potential to occur.
Chloride can influence neuronal activity though. And adult neurons usually have low levels chloride, which makes them responsive to the actions of GABA. But when levels of chloride get too high, the neurons become unresponsive to GABA.
And this is exactly what the researchers found: In the absence of dopamine, the CGIN neurons lost their ability to respond to GABA. And they discovered that this loss of response was due to very high levels of chloride in the cells.
So how does bumetanide correct this?
Chloride is trafficked into and out of cells via chloride channels. NKCC1 is one of those channels.
NKCC1 aids in the active transport of chloride into a cell.
And bumetanide is a NKCC1 chloride importer antagonist – that is a molecule that blocks the activity of NKCC1.

Source: Ahc
By inhibiting the NKCC1 chloride importer, bumetanide efficiently restores low chloride levels in the neuron. This in turn returns the action of GABA to normal, which reduces the level of inhibition in the striatum.
And this effect was demonstrated behaviourally in mouse models of Parkinson’s.
The mice were treated with the neurotoxin 6-OHDA 8 weeks before behaviour testing, and they were treated daily with bumetanide (starting 3 weeks after the neurotoxin was given). These mice could perform the motor tests as well as normal control animals (with an intact dopamine system) and better than untreated mice (with dopamine loss).
To assess motor coordination, the mice firstly had to traverse a rolling beam and the time taken to do this was recorded (graph on the left below). The mice treated with the neurotoxin took longer to complete this task, compared with control animals and mice that were treated with the neurotoxin and bumetanide. Similarly, in a second test of motor ability, the mice had to descend a vertical wooden pole (50 cm long and 1 cm diameter) leading to their home cage. Again the mice treated with the neurotoxin and bumetanide performed better than their untreated counterparts (graph on the right below).

Bumetanide corrects behavioural deficits in mouse model of PD. Source: Nature
These results lead the researchers to conclude that high levels of chloride – causing constant GABA activity in CGINs – contribute to the motor problems produced by dopamine depletion. And this contribution can be reduced by lowering the levels of chloride in the cells via the treatment of the NKCC1 chloride importer, bumetanide.
|
# # # RECAP #3: Bumetanide has been tested in models of Parkinson’s and it exhibited encouraging effects on motor symptoms. It’s effects on APOE4 has not been evaluated in this context. # # # |
Interesting. Has Bumetanide ever been clinically tested in Parkinson’s?
There has been a very small pilot study conducted by the same French research group that produced the study reviewed above.
This is the research report:

Title: Bumetanide to Treat Parkinson Disease: A Report of 4 Cases
Authors: Damier P, Hammond C, Ben-Ari Y
Journal: Clin Neuropharmacol. 2016 Jan-Feb;39(1):57-9
PMID: 26757306
In this open label pilot study, the researchers treated 4 people with Parkinson’s with bumetanide daily for 2 months. They found that bumetanide was safe and well tolerated, and the treatment resulted in an improvement of Parkinson’s motor symptoms in all 4 subjects (as determined by the unified Parkinson’s disease rating scale (or UPDRS) both ON and OFF L-dopa medication). The investigators also reported that bumetanide improved gait and freezing in 2 of those participants.
Here is a table outlining the results in each case:

Source: Frontiersin
Based on all of these results, the researchers set up a Phase II clinical trial to blindly evaluate bumetanide as a potential treatment for Parkinson’s in a cohort of 40 affected individuals (Click here to read more about this study).
They did this via a company called B&A Therapeutics.

Source: B&A Therapeutics
The study was supposed to be completed in September 2020, but I guess COVID may have been a problem. The company website has not been updated for some time, and the ClinicalTrials.gov website has listed the status of this trials as “Unknown”.
So hopefully we will hear some news about this study in the near future.
In September of this year, however, another biotech company – Neurochlore – evaluating bumetanide in the treatment of Autism Spectrum Disorders in children and adolescents announced that the treatment exhibited no sign of effectiveness in their two Phase 3 clinical studies (Click here to read more about this).
Another small study (n=6) conducted in Kuwait found that bumetanide improved freezing of gait issues in PD (Click here to read a conference abstract about this).
Interesting. But if I am an APOE4-carrier, and I am not comfortable about taking medication like bumetanide, is there anything I can do?
There is a great deal of research being conducted on APOE4 at present and earlier this year, this interesting report was published:
 Title: Association of Physical Activity and APOE Genotype With Longitudinal Cognitive Change in Early Parkinson Disease.
Title: Association of Physical Activity and APOE Genotype With Longitudinal Cognitive Change in Early Parkinson Disease.
Authors: Kim R, Park S, Yoo D, Jun JS, Jeon B.
Journal: Neurology. 2021 May 11;96(19):e2429-e2437.
PMID: 33790041
In this study, the researchers want to determine if being more physical active could help to modify the negative association of APOE ε4 with longitudinal cognitive changes in early Parkinson’s. To do this, they used longitudinal self-reported physical activity data from the Parkinson’s Progression Markers Initiative (PPMI study). The data came from the assessments performed at years 2, 3, and 4 of the study. They compared this physical activity data with measures of cognitive function (such as the Montreal Cognitive Assessment (or MoCA)).
They used data from 173 participants with early Parkinson’s (average age was 63 years). Approximately 1/4 of these cases (27%) were APOE4 carriers, and these individuals exhibited a steeper rate of cognitive decline than the rest of the participants.
BUT, the APOE4 carriers that had higher physical activity displayed a statistically significant slower rate of cognitive decline (p = 0.001), indicating that increased physical activity may be able to reduce APOE4-related vulnerability to early cognitive decline in people with PD.
Exercise everyone.
Just do it.
So what does it all mean?
Drug repurposing representing a rapid means of bringing novel therapies to a patient community. Well characterised clinically-available treatments can be tested for safety and efficacy in new medical indications providing a proof-of-principal result that can lead not only to new treatment options, but also new insights into the underlying biology of a condition.
New preclinical research points toward the possibility of repurposing the heart failure treatment bumetanide for APOE4-associated conditions like Alzheimer’s and Parkinson’s, but a lot of research is still required to determine if the treatment could be safe. The initial preclinical results will need to be independently replicated, and then caution will be required in monitoring patient cohorts being administered the treatment. Bumetanide administration can lead to serious water and salt/mineral loss, as well as a reduction in blood pressure – leading to dehydration and dizzy spells (Click here to read more about thoughts on required caution).
APOE4 has a serious genetic association with cognitive decline, however, and it is extremely encouraging that researchers have been able to identify clinically available agents that could potentially be helpful for affected carriers. As with all of the research discussed on this website, it will be interesting to see how this particular story progresses.
 All of the material on this website is licensed under a
All of the material on this website is licensed under a
Creative Commons Attribution 4.0 International License
You can do whatever you like with it!
EDITOR’S NOTE: The information provided by the SoPD website is for information and educational purposes only. Under no circumstances should it ever be considered medical or actionable advice. It is provided by research scientists, not medical practitioners. Any actions taken – based on what has been read on the website – are the sole responsibility of the reader. Any actions being contemplated by readers should firstly be discussed with a qualified healthcare professional who is aware of your medical history. While some of the information discussed in this post may cause concern, please speak with your medical physician before attempting any change in an existing treatment regime.
The banner for today’s post was sourced from drugs